






How do you decide whether to search by protein, cDNA, or genomic position? And how does Mastermind treat those inputs differently? In this blog, we explore strategies for searching single nucleotide variants (SNVs) and ways to get optimal results, written by Mastermind expert Denice Belandres.
Getting Started with Searching SNVs
The Mastermind Genomic Search Engine was built to help genomic scientists save time and never miss a paper by querying the most comprehensive database of genomic evidence. With so much information available behind the search bar, even the most experienced users want to know – how should I craft my search to get optimal results returned? Striking the balance between sensitivity and specificity can be a challenge during literature search. In this post, we’ll dive into strategies for searching single nucleotide variants (SNVs) quickly.
One variant may go by dozens of different names in the literature. Despite HGVS recommendations, a tremendous amount of variation exists in how authors describe genetic variation. What a unique challenge this poses for those of us whose lives revolve around finding evidence in genomic literature! During your routine literature search, you’ve maybe asked yourself…
- Am I thinking about every way an author might describe this variant?
- Does it matter if I search using the three-letter amino acid vs the one-letter?
- What if an author made a typo?
- How do I deal with legacy nomenclature?
- Are there historical shifts in the genome that I don’t know about?
- Has this gene previously gone by a different name?
All of these questions lead to a bigger one: “how sensitive is my literature search engine?” Does it understand the nuances and intricacies baked into how we describe genetic sequence variation? Does it normalize the user input and recognize different ways to talk about the same thing (p.Arg43Ter = Arg43Ter = R43X = c.127C>T = C127T = 127C/T = c.127C→T)? If you’re using Mastermind – you can breathe easy – because we’ve thought about these situations.
Thanks to Mastermind’s Genomic Language Processing (GLP), you don’t have to think about every way an author might describe a variant. When you search a variant one way, Mastermind is looking for matches for all conceivable iterations of that variant. You can see this in action when you perform a variant search in Mastermind, then click the Google Scholar link out near the Articles list. The information shown in the Google Scholar search bar reflects all the different ways you’re querying the Mastermind database, just by searching “BRCA1:p.E1535X”. You would have to know about all of these and type them each into Google Scholar to get the same level of sensitivity out of that search engine.
![]()

The full query sent to Google Scholar by Mastermind: “brca1” ( “brca1” ( “p.Glu1535Ter” | “Glu1535Ter” | “E1535X” | “p.E1535X” | “c.4603G>T” | “4603G>T” | “G4603T” | “4603G/T” ) )
Searching Variants by Genomic Position
Our philosophy has always been to put sensitivity first. After all, you can’t evaluate data that you don’t have. In other words: a false negative is non-recoverable. But this begs the question, how then should I decide between searching my variant as a protein change, cDNA, or a genomic position? Does Mastermind treat those inputs differently? If so, how and why?
In short, yes, the results are different based on how you launch your variant search. The graphic below explains what filtering is happening behind the scenes when a variant is searched.
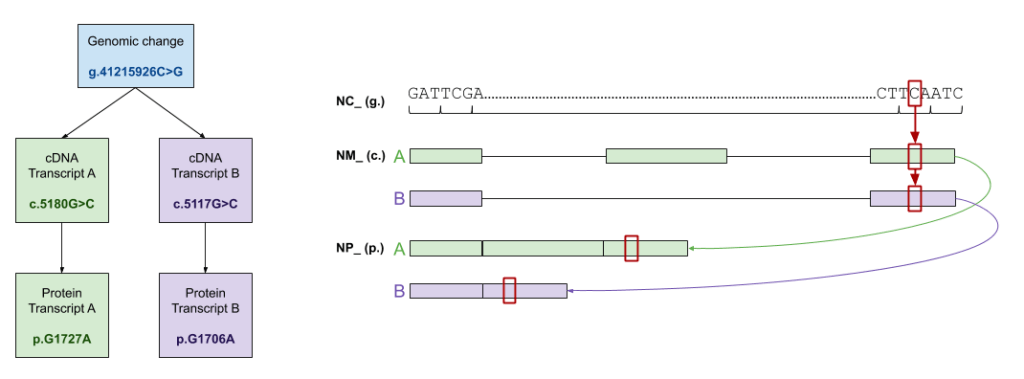
A genomic position search is the least restrictive – all changes at that position are returned in the search. This could include changes on any transcript, and even changes in different genes than the one you might have in mind. Pro tip: add your gene to the search bar if you want to limit results to hits in that gene.
Searching Variants by cDNA Position
A step beyond a genomic position search is a cDNA search. Results are filtered to transcripts where that specific cDNA change is valid. Similarly, with a protein search, results are filtered to transcripts where that specific protein change is valid. Of note, there are instances where multiple cDNA changes can result in the same protein change. Another reason we treat cDNA and protein nomenclature as filters is to avoid bidirectional ambiguity in the results Mastermind returns.
When cDNA nomenclature is detected in an article, you can hover over the match within the full-text matches to see transcript information, as shown below.
![]()
Within the full-text matches, the MATCHED column indicates the exact nomenclature found in the article. Scanning through the sentence fragments allows users to quickly identify whether the variant is a true match or a possible false positive. While false negatives are non-recoverable, false positives are simply addressed by looking at the data.
Legacy numbering, historical shifts, signal peptides, updated gene names – these can all muddy already murky waters. At Genomenon, we’ve indexed over 9 million full-text genomics articles to date. Moreover, our team of clinical genomic scientists spends their days reading these articles, leveraging Mastermind’s sensitivity to manually curate variants. As far as horrible nomenclature goes, we’ve seen it all! And our curation team’s experience gets funneled back to our development team, who are continually improving GLP and how we index articles.
One thing about the Genomenon team: we are always learning. Whether you have a question, some feedback, or a concern – we want to hear from you. Click the Contact Us button within the application, or send us a message: support@genomenon.com
Still need to create your Mastermind account? Click here to sign up and start searching!
ABOUT THE AUTHOR
 Denice Belandres, Field Application Scientist, Genomenon
Denice Belandres, Field Application Scientist, Genomenon
Denice provides technical support and training to Mastermind users at all levels. With a background in germline variant analysis and preimplantation genetics in clinical NGS labs, she turns feedback into function, enabling implementation of Mastermind for a variety of clinical use-cases.
Additional reading: Discrepancies in Variant Classifications and How to Resolve Them
- Am I thinking about every way an author might describe this variant?
- Does it matter if I search using the three-letter amino acid vs the one-letter?
- What if an author made a typo?
- How do I deal with legacy nomenclature?
- Are there historical shifts in the genome that I don’t know about?
- Has this gene previously gone by a different name?
Explore & Discover!
The Genomenon Team
Learn more about Genomenon at genomenon.com and follow them on LinkedIn and Twitter.





